


Discover more from Who is Robert Malone
A (Draft) Blueprint for the Reorganization of the FDA


An (Initial Draft) Blueprint for the Reorganization of the FDA
By:
Robert W. Malone, MD, MS
Jill G. Malone, PhD
Written for the Make America Healthy Again coalition.
This initial draft blueprint is not a “final” version and represents only a first draft and initial concept paper of some of the major structural changes that must occur at the FDA.
Disclaimer: All opinions in this draft blueprint are the author’s alone, and all errors in judgment are also theirs. The authors do not represent any organization or company, including MAHA.
The content is provided as a video and text following the video below.
Executive Summary
The election of President Donald Trump has provided a unique mandate and opportunity for implementing widespread reorganization and change in federal government structure, emphasis, and mandates to more closely align with original constitutional intent and general citizen interests – with an unapologetic focus on USA national interests in both domestic and foreign affairs. In other words, an emphasis on revitalizing the USA and reimagining the role and scope of the Federal Republic, making America great again.
Robert F. Kennedy Jr. and the Make America Healthy Again initiative have committed to completely overhauling those agencies involved in our nation's health, from the science and medicine to the foods and drugs we eat.
The MAHA agenda includes combating the chronic disease epidemic by addressing the widespread health issues affecting Americans, supporting regenerative farms and the small farmer to both rebuild America to create healthier humans and dismantling the ecosystem of corporate corruption and agency capture which have evolved to characterize the US Federal bureaucratic system.
The following draft text introduces many institutional issues (and potential solution strategies) which may strengthen the core mission of the FDA and bring it more into alignment with a focus on making America healthy again.
To make these changes appropriately, rapidly, and maintain compliance with the scope of the law, the original Acts of Congress must be considered in any regulatory process and institutional changes. Therefore, provided is a list of key congressional Acts that have both chartered the FDA and been used to modify and clarify its mission and responsibilities over time.
A Blueprint for the Reorganization of the FDA:
Directives, Policies, and Procedures
The FDA regulates food and drugs, and has significant responsibilities to safeguard the health of the American public. Therefore, any changes have to be performed with thoughtful, transparent, and deliberate methodology. Unfortunately, the experience of executive and legislative branches over time has often been that the FDA is aloof, unresponsive, non-transparent and frequently appears to be passive aggressive when responding the requests and reform-oriented initiatives.
As a component of Trump’s and MAHA’s overall broad policy agenda to change the administrative policies and organizational cultures of agencies like the FDA, NIH, CDC, USDA and more broadly HHS – and eliminate regulatory capture -- President Trump and the intragovernmental MAHA initiative might consider instituting some or all of the following changes at the FDA.
Broadly speaking, potential policy initiatives include both modest internal FDA policy changes (for example involving packaging and labeling requirements), technology updates (focused agency implementation and integration of both artificial intelligence and updated statistical analysis methods) as well as broad based, whole of government initiatives. One example to illustrate opportunities in breaking down intragovernmental silos involves integrating epidemiological and adverse event reporting systems from Medicare, Medicaid, the Veterans Administration, the DoD healthcare system, the Indian Health Service, the Centers for Disease Control and Prevention, and (ideally) private sector health systems. In the case of a broad database/data fusion and statistical initiative, such an initiative might eventually integrate health and adverse event-related data from willing and qualified allied nation-states.
On an even broader horizon, the FDA in cooperation with other agencies might play a key role in reimagining the entire structure of federal Research and Development investments, policies and structures as an update to the current system, which is based on the post WWII evaluation and proposals of Vannevar Bush during his service as the first Presidential Science Advisor. It is time to revitalize the federal research and development enterprise to reduce waste, reconsider the role of academia, facilitate innovation (as defined by making America healthy again), and provide greater return on investment to the taxpayers.
High level initiative options:
President Trump may consider banning prescription drug ads via an executive order. Almost all other nations in the world do not allow such advertising. However, this is a short-term fix. The example of how Congress, beginning in 1970, banned tobacco ads shows that this can be accomplished. The commissioner of the FDA might prioritize getting similar legislation passed through congress.
FDA personnel should be reorganized and refocused on the FDA core mission, with conflicts of interest clearly identified and eliminated. There should be better firewalls implemented to separate pharmaceutical industry interests from FDA oversight functions.
The history of collusion between different HHS senior leaders to circumvent the specific will of the President to provide access to a particular FDA-authorized medicines (such as HCQ) to US Citizens demonstrates the need to enhance Presidential authority to fire and replace Senior Executive Service and GS-rank personnel. With an executive order reinstating “schedule F” or similar, combined with solid leadership at the FDA, the proper function of the FDA relative to the executive leader of the United States, which requires efficient and responsible responsiveness to the sitting president, can be restored.
The FDA should not be allowed to choose winners and losers in terms of either specific technologies, pharmaceutical companies, or medical products. Such policies must be codified in the form of revised administrative policies, reinforced by clear disciplinary practices, and policed by both systematic internal audits and specifically appointed and empowered oversight and ombudsman personnel and policies.
The ill-advised legislative decision to partially fund the FDA via industry fees has created an unintended intrinsic conflict of interest on the part of the FDA as an organization as well as its employees. The public may save some taxpayer dollars through this arrangement, but it has been ill-served consequent to the resulting conflicts of interest in which the FDA has become dependent on industry funds. This includes the revolving door between personnel who have approved clinical trial results and products and those with subsequent employment within that same company. A prime example of this practice is Dr. Scott Gottleib, who within a year of stepping down from this position as Commieoner at the FDA joined the board of Pfizer. This practice must stop.
As the user fee program was mandated by Congress, first in 1992 and then re-authorized through 2027, initial changes to the program might include the placement of firewalls between pharma and medical device companies, as well as the personnel whose paychecks they are funding. Stricter contracts regarding future employment after working at the FDA should be established.
User fee calculations and processes must be firewalled from FDA clinical trial regulators.
Regulatory positions and policies of the FDA must become aligned with the recent Supreme Court decision overturning the Chevron Deference. With support from appropriately trained Artificial Intelligence algorithms, all FDA regulatory positions must be reviewed to determine whether they represent unilateral administrative decisions (regulatory mission creep and overreach) or are firmly based on congressional authorization statute language. Those policies determined to be non-compliant with congressional authorization must be promptly rescinded.
The Emergency Use Authorization (EUA) process and authority must be reviewed and revised to identify and resolve intrinsic conflicts, contradictions, and implementation ambiguity. This includes compliance with the letter of the law relating to informed consent under EUA. Terms and conditions within current EUA guidance concerning patient-informed consent will be reviewed and revised to ensure compliance with Belmont report findings and Common Rule federal law to strengthen transparency while preserving and better protecting patient rights for full and open informed consent, coupled with explicit prohibitions on enticement, coercion and compulsion to accept EUA products. Whether authorized under EUA or more traditional market authorization policies, all federal communication relating to FDA authorized products must be in full compliance with the scope and specifics of that authorization and shall not exceed authorized indications.
A formal, fully independent “lessons learned” assessment of FDA successes, inefficiencies, and failures during the COVID crisis and Emergency Use Authorization processes must be performed with specific actions proposed and implemented to address identified strengths and weaknesses. Assessment of independence and potential sources of conflicts of interest in external FDA advisory committees such as the FDA’s Vaccines and Related Biological Products Advisory Committee will be performed, and more stringent policies and practices implemented.
The emergency use authorization for the Pfizer and Moderna mRNA COVID-19 vaccines should be terminated immediately via executive order. FDA should require that these and related vaccine products be more appropriately classified as gene therapy-related products and must meet both vaccine-related and gene therapy product-related guidance including rigorous assessment of genotoxicity, shedding, biodistribution, fetal/pregnancy and other toxicology risks. Rigorous, well-controlled long term safety studies must be required before considering future market authorization for these or other RNA, DNA, or other gene therapy-related products including self-replicating RNA-based products.
President Trump and his administration should consider developing recommendations for congressional actions to revise existing legislation (Pandemic and All Hazards Preparedness Act, Public Readiness and Preparedness Act, Coronavirus Aid, Relief and Economic Security Act, etc.) to improve administrative FDA responsiveness to POTUS and public needs, and to facilitate greater cooperation with practicing frontline physicians during future public health crises.
The physician’s right to prescribe off-label drugs must be preserved and strengthened.
Marketing, labeling and packaging guidance should be revised with a focus on fostering and enabling informed consent of the general public regarding the risks and benefits of all FDA regulated products. As is the norm with clinical research informed consent, package insert and all other labeling and marketing information must be carefully reviewed to insure that key information is readily understood by the intended consumer. For example, greater use of illustrations, language not to exceed ninth-grade reading competency standards, and translations for those non-native speakers targeted in packaging, advertising and marketing materials should be considered.
Strong firewalls shall be developed to separate the regulatory oversight activities of the FDA from interference from other federal stakeholders including CIA/DARPA, DoD, NIH, and CDC.
The issue of royalties obtained by inventors whose names are on patents generated from inventions funded by the federal government must be addressed. However, the Bayh-Dole Act, which defines this policy, can only be modified by Congress. The unintended consequence of the Bayh-Dole Act is that federal employees who can benefit from royalty payments have a conflict of interest in directing funds, prioritizing clinical trials, and policies that benefit that employee directly via royalty payments. Those payments may be equal to or exceed that employee’s salary.
But there is a way to circumvent the conflicts of interest that royalty payments bring. The Bayh-Dole Act does not provide a uniform percentage royalty for federal employees. Instead, it establishes a framework for negotiating royalty rates on a case-by-case basis. The solution, then, is simple. Each FDA employee must sign a statement at the time of employment stating that their employment compensation is adequate payment for any inventions patented under the terms of the Bayh-Dole Act while in the employment of the federal government. Current employees must sign this agreement retroactively. Any employees receiving royalties or whose names have been put on patents or patent applications must be removed from any and all projects, clinical trials, and approval processes related to the invention(s).
All DEI hires and promotions must be re-evaluated. All current and future employees and their promotion must be based on merit and job/task-oriented competency.
All work-at-home policies and practices must be reevaluated with an emphasis on on-site work practices.
Environmental, social, and governance (ESG) is shorthand for an investing principle that prioritizes environmental issues, social issues, and corporate governance. Global change and green regulations have been written into almost all federal agency regulatory codes. ESG has been inserted into many FDA programs to evaluate government contracts, grants for biologics, drug development, new medical devices, and food safety. Climate change initiatives should not be within the FDA’s purview. That includes food supply grants, contracts, projects, research, and public outreach.
The concept of One Health is to optimize the health of people, animals, and ecosystems in a sustainable and balanced manner by recognizing the interconnectedness and interdependence of human health, animal health (both domestic and wild), plant health, and the broader environment, including ecosystems. This flawed concept has evolved into a philosophy that places animals and the environment on par with humans and human health. This has led to disastrous consequences worldwide. One Health has been used by the globalists, WEF, WHO and the UN to expand control and has come to represent the idea that the earth, including the weather, can be controlled. This includes worldwide surveillance programs.
The FDA has endorsed the One Health vision statement, which is “One Health (formerly called One Medicine) is dedicated to improving the lives of all species—human and animal—through the integration of human medicine, veterinary medicine, and environmental science” This essentially puts animal lives on par with human lives. This is an example of mission creep and is outside of the scoop of the FDA’s core mission.
FDA mission creep must end. The FDA’s mission, as per its 1938 congressional mandate, is threefold. The FDA ensures the safety of foods, cosmetics, and radiation-emitting products, regulating the safety and effectiveness of drugs, biologics (e.g., vaccines), and medical devices.
The FDA should systematically eliminate programs and initiatives outside of scope, such as DEI and climate change initiatives as well as eliminate waste. Multiple teams can work independently to eliminate and streamline rules and regulatory processes to avoid groupthink. Teams will go through and strike out superfluous regulations and requirements or ones outside the FDA’s scope. Once this work is finished, the leaders of these teams will be asked to come together to put forth a final document incorporating the work of the independent reports. This model was first used to create the Marshall Plan to rebuild Europe after WWII. It is a tried and true method to eliminate groupthink from planning and strategic processes, and is the recommended approach for developing large government policy initiatives consequent to the case study analyses of Irving Janus as summarized in his seminal work “Victims of Groupthink”.
The FDA must cooperate and work with the new federal Efficiency Department (DOGE) to eliminate waste, unnecessary and illegal regulatory burdens. An AI-driven approach to facilitating this is outlined above. As discussed, FDA policies and regulations need to be reviewed systemically in light of the overturning of the Chevron deference. This is both to bring the FDA back to the roots of this country – as a constitutional republic- and to protect the FDA against future lawsuits and proactively reduce avoidable legal distractions from core missions.
The revolving door between the pharmaceutical, biotech, medical device, diagnostics companies, big tech companies, and the FDA must end. Stronger rules and regulations regarding industry leaders stepping into roles within the FDA and vice versa must be implemented and enforced. FDA employees must be limited in their ability and disincentivized to step into roles within industries that they have regulated.
The FDA has been tasked with regulating food and drugs as well as promoting innovation by Congress. This has created an inherent conflict of interest within the agency because of how innovation has been defined by the FDA. To alleviate this COI, the FDA must find ways to split the two conflicting mandates. FDA departments and offices that regulate specific food and drugs should be completely firewalled from those that promote innovation.
Innovation can be defined differently than it is now. For instance, the FDA could innovate ways to increase wellness instead of promoting the innovation of new biotechnologies. As an example, innovating methods to increase nutritional awareness and decrease low-nutritional value foods in the American diet would be very different endpoints than what the FDA currently defines as innovation.
Staffing issues continue to haunt the FDA. Due to purported staffing shortages, the FDA has a massive backlog of manufacturing inspections of pharmaceutical plants, with an estimated 42% overdue by over five years. The Inspection of clinical trial sites are down 45% since 2019. Approximately 185 factories in China are currently overdue for inspection, and there are only two full-time FDA inspectors stationed in China, complicating the inspection process.
In 2024, Rep. Mike Waltz wrote a letter to the FDA about the issues of telework employees and the fact that a large percentage (unknown) still need to return to the workplace, which has impeded clinical trials and study results.
On November 14, 2024, the Government Accountability Office (GAO) issued a 67-page report that implores the FDA to implement more aggressive steps to attract and retain inspectors. The “GAO recommends that FDA collaborate to develop and implement action plans to address the remaining root causes of investigator attrition that balance inspection needs against the need to retain investigators. HHS agreed with this recommendation.”
The cozy relationship between the FDA and the companies it regulates continues to be an issue. One such example is the relationship between Moderna and the FDA, as outlined by the highly regarded, peer-reviewed medical journal BMJ.
The restructuring of hiring processes, merit-based retention programs, and employee culture is long overdue. Furthermore, inspection processes of manufacturing plants must be more rigorous, with employees not worrying about being punished if they issue a negative review.
Repatriating generic pharmaceutical manufacturing domestically for essential medicines is critical for the health and safety of the nation. The FDA must work with other agencies to ensure that this occurs in an expedited manner.
Repatriating drug supply and pharmaceutical precursor supply chains from “frenemy” (India) or hostile competitor nations (China) is a national security requirement. This must be enabled, facilitated, and expedited by the FDA.
Pre-clinical and clinical trial data evaluated by the FDA must be made available to the public. The clinicaltrials.gov site does require investigators to release some summary data eventually. However, it is often years after the clinical trial has ended. The FDA rarely releases pre-clinical and clinical data. For instance, the mRNA vaccine Pfizer pre-clinical and clinical trial data required a judge’s court order to be released, and then it was under significant duress. Such data should be available to the public, independently and objectively summarized in readily understood language and figures, with a specific objective of facilitating and enabling both informed consent and medical care provider access.
The FDA Adverse Event Reporting System (FAERS) for reported drug adverse events is a “is a highly interactive web-based tool that will allow for the querying of FAERS data in a user friendly fashion. The intention of this tool is to expand access of FAERS data to the general public to search for information related to human adverse events reported to the FDA by the pharmaceutical industry, healthcare providers and consumers.” The event reporting system and dashboard are much more user-friendly than the CDC VAERS system for reporting vaccine adverse events. A re-organization of HHS might include merging the two systems into the FAERS system under the FDA.
A long-term goal would be to use the FAERS system to incorporate other databases such as the Medicare/Medicaid, VA, and DoD databases (DMED and DMSS) for a whole of government database on adverse events reporting.
The FDA needs to investigate both long and short term drug-drug interactions before market authorization. As a specific example and priority, studies must be required to demonstrate that the current federally-recommend pediatric (and legal immigrant) vaccine schedule involving closely spaced dosing of a wide range of different pro-inflammatory vaccine products is safe and effective. Market authorization and dosing recommendations must account for potential drug-drug or biological product interactions between authorized products likely to be co-administered or functionally concurrently administered.
The Human Foods Program (HFP)
The Reagan-Udall Foundation for the Food and Drug Administration is an independent nonprofit organization created by Congress to support and advance the mission of the U.S. Food and Drug Administration (FDA)
In a Reagan-Udall Foundation for the FDA report, published in 2023 titled: Operational Evaluation of the FDA Human Foods Program, is the basis for what the FDA calls a total reorganization of the FDA. In this report, the idea was floated of separating into two separate administrations under the Cabinet-level Director of Health and Human Services. The two programs were proposed to be named the Federal Drug Administration and the Federal Food Administration.
There is merit to the idea as defined in the Reagan-Udall Foundation chart below. But it most likely would require the passage of a Congressional Act.
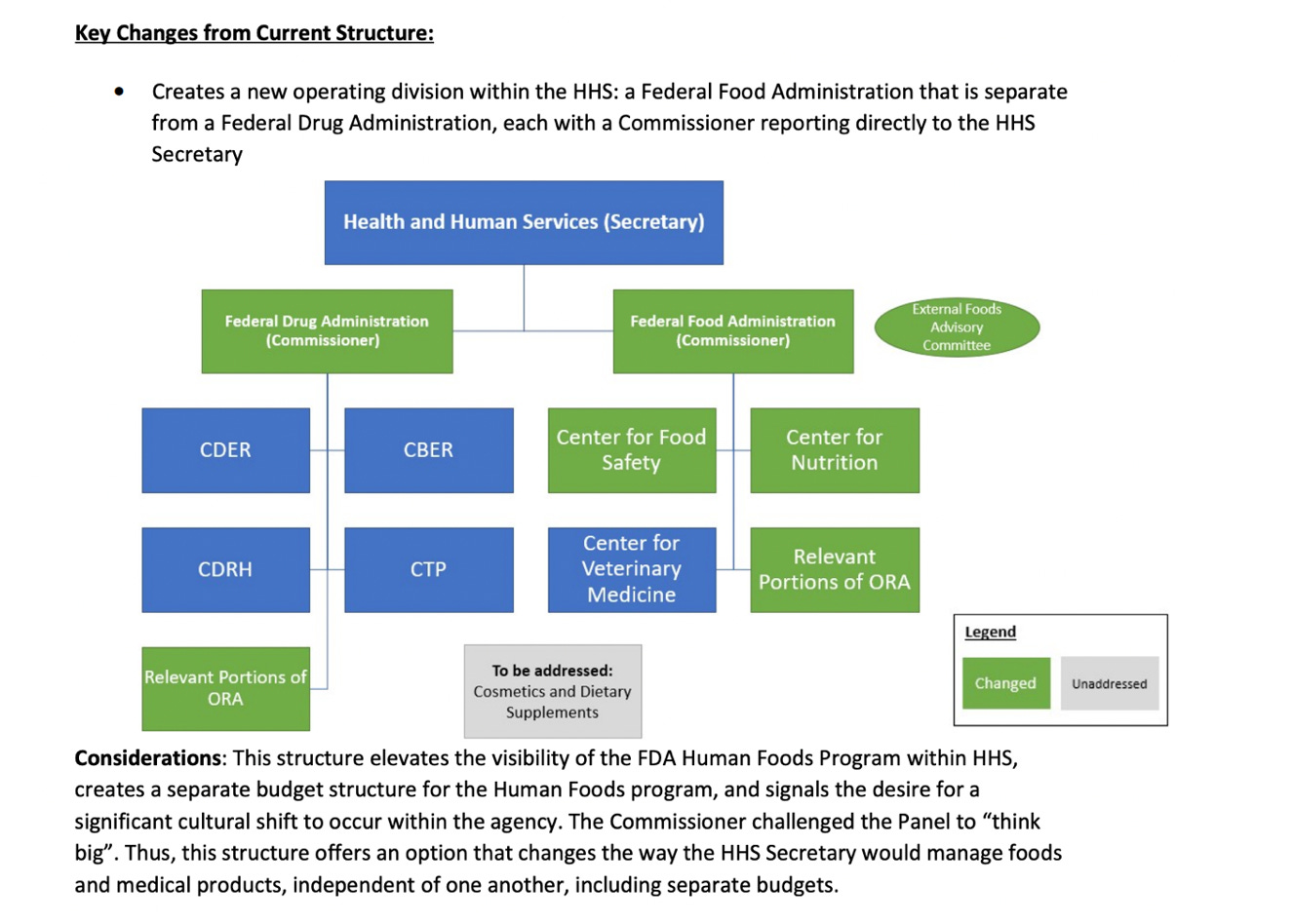
In lieu of the above, the FDA developed The Human Foods Program (HFP) to oversee all FDA activities related to food safety and nutrition, organized in a single group working under the Deputy Commissioner of Human Foods. There has been little public exposure or media attention to what the FDA calls a “major reorganization.”
The program has been defined by the FDA as a centralization of three risk management activities. Those being;
“Microbiological Food Safety: Advancing strategies to prevent pathogen-related foodborne illness in close collaboration with other regulatory agencies, states, industry, and other stakeholders.
Food Chemical Safety: Ensuring that exposure to chemicals, including both additives and contaminants, that occur in foods is safe, advancing dietary supplement safety, and supporting and effectively regulating food ingredient innovation.
Nutrition: Elevating and empowering action on nutrition science, policy, and initiatives to help reduce the burden of diet-related chronic diseases, improve health equity, and ensure the nutritional adequacy and safety of infant formula.”
Once aspect of this program is little more than window dressing for a programmatic initiative to exert more regulatory control on small farmers and producers through contact and genetics testing of food products. The push for more regulations often comes through lobbying organizations working on behalf of large agriculture and processing companies. More regulations are a way of driving small farms and small companies out of business. This is a ploy used by big businesses to control competition.
Through the regulation of pasteurization and interstate commerce, combined with aggressive DoJ action, the FDA stymied direct sales of milk (farm to consumer) over fifty years ago to stop interstate sales of raw milk. These policies still need to be re-examined. Such regulations need to be re-evaluated, given modern husbandry practices – combined with the use of herd testing.
The FDA Food Program also lists chronic disease as a significant concern, which is excellent. However, the website description of the program then combines health equity and baby formula quality and supply to imply that chronic diseases are on par with one another. The inclusion of DEI policies in the objectives of HFP makes the FDA’s commitment to chronic diseases suspect.
Coincidently, the FDA’s reorganization, which started in the fall of 2024, has already developed a framework relevant to the MAHA agenda. The MAHA agenda includes 1) combating the chronic disease epidemic by addressing the widespread health issues affecting Americans, 2) supporting regenerative farming practices and small farms to build health in America, and 3) Dismantling Corporate Corruption and Agency Capture.
By using this new Human Foods Program (HFP) infrastructure, which has not yet solidified into bureaucratic sludge, the Trump/ MAHA administration can instigate rapid infrastructure changes – by simply inserting new programmatic goals for the ones that have only been implemented in 2024, which have not yet had time to harden within the framework of the administrative state.
The issue of chronic diseases and nutrition is something the FDA must have a hand in regulating. For instance, overusing glyphosate in agriculture and as a desiccant for drying grains and legumes, ultra-processed foods, trans fats, and food additives – including preservatives, artificial dyes and colorants all need more oversight.
Key FDA Congressional Acts and Mandates
· The Federal Food, Drug, and Cosmetic Act (1938) and its subsequent amendments which provide the FDA’s statutory authority to regulate food, drugs, cosmetics, and medical devices.
o The Federal Food, Drug, and Cosmetic Act of 1938 is the foundational law that established the modern regulatory framework for foods, drugs, medical devices, and cosmetics.
o Mandates pre-market approval of new drugs.
o Prohibits false therapeutic claims for drugs.
o Set standards for food safety and labeling.
· The 1958 Food Additives Amendment requires manufacturers to establish the safety of new food additives before going to market.
· Kefauver-Harris Amendment of 1962. This amendment strengthened drug regulations by requiring manufacturers to provide evidence of a drug's effectiveness before approval, mandated adverse event reporting, and established good manufacturing practices (GMP).
· The 1980 Infant Formula Act requires manufacturers to follow quality, nutrient, and stability protocols.
· Orphan Drug Act of 1983. This act provides incentives for developing drugs to treat rare diseases, including tax credits, as well as extends market exclusivity.
· The 1990 Nutrition Labeling and Education Act requires nutrition labels and regulates health and nutrient claims.
· National Childhood Vaccine Injury Act of 1986 {CRS, 1986 #1}. This act established the Vaccine Adverse Event Reporting System (VAERS) for monitoring vaccine safety. It also created a no-fault compensation system for vaccine-related injuries.
o The summary text of the bill states that it “Amends the Public Health Service Act to establish in the Department of Health and Human Services a National Vaccine Program to: (1) direct vaccine research and development within the Federal Government; (2) ensure the production and procurement of safe and effective vaccines; (3) direct the distribution and use of vaccines; and (4) coordinate governmental and nongovernmental activities. Requires the Director of the Program to report to specified congressional committees.
o Established the National Vaccine Advisory Committee at the CDC to recommend: (1) ways to encourage the availability of an adequate supply of vaccines; and (2) research priorities.
· Prescription Drug User Fee Act (PDUFA) of 1992, with its subsequent reauthorizations, established user fee programs for pharmaceutical and biologic products – which allowed for expedited reviews.
The FY 2025 PDUFA fee rates were published in the Federal Register (FR) on July 31, 2024. For more information regarding the FY 2025 fee rates, please see the FR notice.
On September 30, 2022, President Biden signed into law the FDA User Fee Reauthorization Act of 2022. This includes the reauthorization of the Prescription Drug User Fee Act (PDUFA VII) from fiscal year (FY) 2023 through 2027. Additional information can be found on the PDUFA VII web page.
· The 1994 Dietary Supplement Health and Education Act created a new regulatory framework for dietary supplements
· The 1997 Food and Drug Administration Modernization Act. This act modernized the regulation of food, drugs, devices, and cosmetics. It includes provisions for the Fast track approval of drugs for serious conditions, expanded access to experimental drugs and regulation of health claims for foods.
· The 2004 Food Allergen Labeling and Consumer Protection Act requires food labels to include key allergens.
· The 21st Century Cures Act (2016). This Act aimed to accelerate medical product development to bring new innovations to market faster and more efficiently. This Act has benefited the pharmaceutical, medical device, and biologics industries.
o This act aimed to accelerate medical product development and bring innovations to patients faster. Key provisions include:
o Establishing new expedited product development programs
o Incorporating patient perspectives into the drug development and approval process
o Modernizing clinical trial designs and assessments
· FDA Reauthorization Act of 2017 (FDARA)
o This act reauthorized several user fee programs and included provisions to:
§ Enhance FDA's ability to use real-world evidence
§ Improve generic drug development and review
§ Facilitate pediatric drug development
o The Act specifically focuses on several key areas:
Accelerating research and drug development
Improving the FDA approval process
Enhancing data sharing and electronic health records
Addressing mental health and substance abuse issues
Advancing precision medicine and biomedical research
Funding and Initiatives
The Act authorized $6.3 billion in funding, primarily allocated to:
The National Institutes of Health (NIH) for various research initiatives
The Food and Drug Administration (FDA) to streamline drug and device approvals
States to combat the opioid epidemic
Specific initiatives funded include:
The Cancer Moonshot
The BRAIN Initiative (Brain Research through Advancing Innovative Neurotechnologies)
The Precision Medicine Initiative
The Regenerative Medicine Innovation Project
Changes to FDA Approval Process
The Act modifies the FDA drug approval process by:
· Allowing for "data summaries" and "real world evidence" in certain cases rather than full clinical trial results
· Facilitating the development and approval of drugs for rare diseases
· Creating new expedited product development programs, such as the Regenerative Medicine Advanced Therapy (RMAT) and the Breakthrough Devices program
· Health Information Technology
o The Act aims to improve the use of electronic health records and promote interoperability by:
o Addressing information blocking
o Requiring the adoption of standardized processes for exchanging information
o Updating requirements for the ONC health IT certification program
Mental Health and Substance Abuse
The Act allocates $1 billion in grants to states over two years to fight the opioid epidemic, allowing for:
Improved prescription drug monitoring programs
Increased accessibility to treatment programs
Training for healthcare professionals in addiction treatment
Research on effective approaches to prevent dependency
The Responsibilities of the FDA Have Been Defined As:
1. Enforcement of Laws: The FDA is responsible for enforcing laws enacted by Congress, including the Federal Food, Drug, and Cosmetic Act of 1938 (FD&C Act). The goal is to protect the public health and safety.
2. Regulatory Authority: The FDA has regulatory authority over various industries, including pharmaceuticals, medical devices, biologics, animal drugs, tobacco products, cosmetics, and food, to ensure their safety, efficacy, and quality.
3. User Fee Programs: The FDA collects user fees from manufacturers to expedite the review of new drug applications (NDAs) and other product submissions, as authorized by the Prescription Drug User Fee Act (PDUFA) and other user fee programs.
4. Legislative Affairs: The FDA’s Office of Legislation advises and assists Members of Congress and congressional committees on agency actions, policies, and issues related to legislation that may affect FDA, ensuring accurate and timely information exchange.
5. Compliance and Enforcement: The FDA is responsible for ensuring compliance with federal laws and regulations. This includes inspections, investigations, and enforcement actions.
6. Promotion of innovation in food safety and drugs.
**************
The authors have prepared this analysis in the hope that it will help guide the transition team and new HHS leaders. Of course, as time was limited and this is just a draft - a more in-depth analysis is already being developed.
**************
Consider a subscription to Malone News to help change Americans' hearts and minds and get the truth out beyond our own echo chamber.
Each day we reach hundreds of thousands of people - but it takes time, money, and resources.
Every bit helps!
This draft makes me very excited to see started. You are in a unique position, with a wide range of understanding of these processes, as well as an ear to Kennedy and others who know and respect both of you. Full speed ahead!
The key issue is the fact that Big Pharma pays user fees to the FDA and the money is ear marked for the exclusive use of processing NDA's for approval. Over 50% of FDA annual budget is ear marked this way. The solution is to take most of the NAIAD budget and transfer it to the FDA and have the FDA manpower be redirected to doing what it's original mission was. Protect American Citizens from harm. From Food, Drugs, Vaccines and Cosmetics that HARM! Second, the revolving door between FDA and Big Pharma has to be locked. Most ex FDA commissioners have gone over to the dark side. Dr. Kessler is one that has not. Big Tobacco tried to black ball him because he and Dr. Koop believed nicotine cigarettes should be managed as a drug. In 2025, the fight to return Americans into healthy lifestyles will be enormous.