

Is the Current Bird Flu Made in America?
Does a new publication in "Poultry, Fisheries & Wildlife Sciences" make this claim?


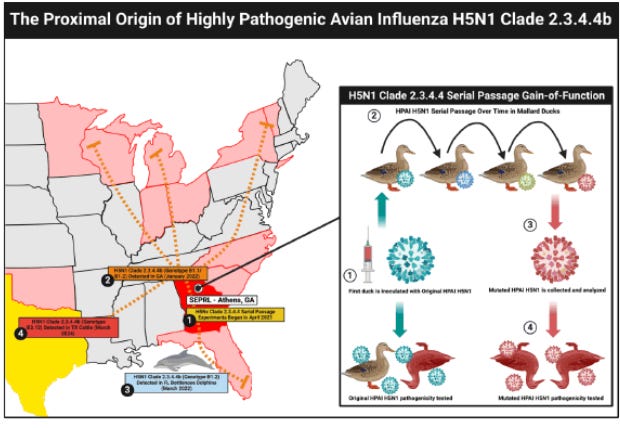

On social media and in broadcast interviews, very strong statements about the origin of the currently circulating Avian Influenza are being made by a team comprised of a clinical renal cardiologist, a writer of true crime novels, and an MPH who graduated in 2024 with a specialization in epidemiology. None of these authors has any training or experience in virology, sequence analysis, viral evolution or geospatial tracking of viral evolution. If true, the assertions that the H5N1 strain currently circulating in the United States were created in an Athens, Georgia, USDA laboratory and (simultaneously, independently?) in the Rotterdam, Netherlands Erasmus medical center would be explosive, would increase general public fear, and would certainly increase distrust of both USDA and other US Government-sponsored virology research. If false, this would be another clear-cut example of psychological bioterrorism, particularly if any associated conflicts of interest were identified..
Dr. Peter McCullough: "The current strain of bird flu is a product of gain of function research done in the USDA Poultry Research Laboratory in Athens, Georgia. So it is a man made problem that, our farms are experiencing right now. It's in the peer reviewed literature, and, I mean, it's really you know, the next steps in this outbreak is, for people to understand, you know, the ramifications of it personally and how to be prepared." "It's the circulating clade, which is the kind of the original source strain, is clade 2.3.4.4b, and, again, that is a product of what's called serial passage gain of function research done at the USDA Poultry Research Laboratory." "It was assisted, by University of Wisconsin School of Veterinary Medicine and, Rotterdam University. The gain of function was to get it to spread from chickens into migratory waterfowl or mallard ducks. And so that's how it's spreading across the world now." "It's been spreading for 4 years. It continually reinfects the farms because the mallard ducks fly around, and they land in in ponds on farms and they, easily infect the other animals, on the farm. So it was able to, in a sense, expand the host range even into cattle and to sea mammals." "Now the good news is it's much milder than bird flu of decades ago, and then there's the peer reviewed paper by Nick Hulscher on this. And, you know, and this has not been denied by the USDA Poultry Research Center." "McCullough Foundation attended the bird flu summits in both, University of Arkansas as well as in Washington, DC. So there's no denial of this. Just like COVID 19, the bird flu problem is a man made problem by the US government."
From the PsyWar Glossary:
Psychological Bioterrorism is the use of fear about a disease to manipulate individuals or populations by governments and other organizations, such as Big Pharma. Although the fear of infectious disease is an obvious example, it is not the only way psychological bioterrorism is used. Other examples include propaganda regarding environmental toxins, unsafe drinking water, soil contamination, and climate change risks. Another name for psychological bioterrorism is information bioterrorism.
The publication received an extremely rapid review and was then promptly published— less than one month between assignment to an editor and actual publication date. As someone who has reviewed many papers and served as editor in chief of a vaccine journal, this is highly unusual and suggests a risk that the review was cursory.
Received: 03-Oct-2024, Manuscript No. PFW-24-34400; Editor assigned: 07-Oct-2024, Pre QC No. PFW-24-34400 (PQ); Reviewed: 21-Oct-2024, QC No. PFW-24-34400; Revised: 28-Oct-2024, Manuscript No. PFW-24-34400 (R); Published: 04-Nov-2024 , DOI: 10.35248/2375-446X.24.12.286
Key elements of this recent publication in question include:
Abstract
“Significant mutations found in recent human cases suggest possible links to serial passage experiments. However, causation has not been established, and further investigation is urgently needed to confirm these findings and to identify all H5N1 laboratory leaks that may have occurred with a focus on mallard ducks and other migratory waterfowl, which have the potential to infect a large number of poultry and livestock facilities around the world.”
Conclusion
“The proximal origins of highly pathogenic avian influenza H5N1 Clade 2.3.4.4b may be the USDA Southeast Poultry Research Laboratory (SEPRL) in Athens, Georgia and the Erasmus Medical Centre in Rotterdam, the Netherlands. Genetic evidence and historical context suggest that laboratory activities, including serial passage and GOF research, could have contributed to the emergence of H5N1 clade 2.3.4.4b. However, causation has not been established, and further investigation is urgently needed to confirm these findings and to identify all H5N1 laboratory leaks that may have occurred with a focus on mallard ducks and other migratory waterfowl, which have the potential to infect a large number of poultry and livestock facilities around the world.
Note the caveats - in no way does this paper actually “prove” anything. At best, what are presented are speculative and rather alarming hypotheses. Origin may be… Suggest that laboratory activities… could have contributed… causation has not been established.
This is not a rigorous scientific study, it is a hypothesis paper. It proves nothing. There are many alternative hypotheses for the origin of the current genotype and phenotype of the currently circulating H5N1 strain. This is one of those many, and as such certainly merits some investigation, but in no way has this manuscript proven the authors’ hypothesis.
In no way are any of these authors “top scientists” with expertise in this area.
To illustrate the level of detail and analysis required to perform a rigorous cladistic, geospatial analysis of viral evolution (which is basically what these three authors claim to have performed, whether or not they know it), please see this example:

This should establish my bonafides regarding this type of research for any skeptics. I am a trained, highly published, and highly cited molecular biologist and virologist. My Google Scholar rankings in these and related areas are equivalent to those of a senior full professor.
Here is one example of how cladistic geospatial tracking has been used by experts to track H5N1 evolution:

This is highly specialized, technically demanding, and computation-intensive work. Not something that a team composed of a renal cardiologist, a true crime writer and a recently minted MPH/epidemiologist are trained or qualified to do.
Are there any relevant Conflicts of Interest (COI) disclosed or otherwise involved in this publication?
These days, most journals require a rigorous and complete disclosure of potential conflicts of interest. No formal COI section or declaration is provided. All three authors are listed as affiliated with the McCullough Foundation.
Neither listed nor disclosed is that the senior author is the chairperson of the scientific board and a key employee of The Wellness Company. The Wellness Company markets non-FDA authorized pharmaceutical products and “Contagion Emergency kits” apparently designed by the senior author and claimed to mitigate the effects of infection by a variety of viruses. At a minimum, this appears to be a financial conflict of interest on the part of the senior author.
Are there recent review publications also focusing on the origins of H5N1 clade 2.3.4.4?
Why yes, there are. Here is one from the American Society of Microbiology:

Here is the conflict of interest statement from this particular review article, to illustrate modern standards for COI disclosure:


What is the actual history of Highly Pathogenic Avian Influenza Virus across the world and in the USA?
Quoting from “Avian influenza A (H5N1) virus in dairy cattle: origin, evolution, and cross-species transmission.” For corresponding references, see this linked publication.
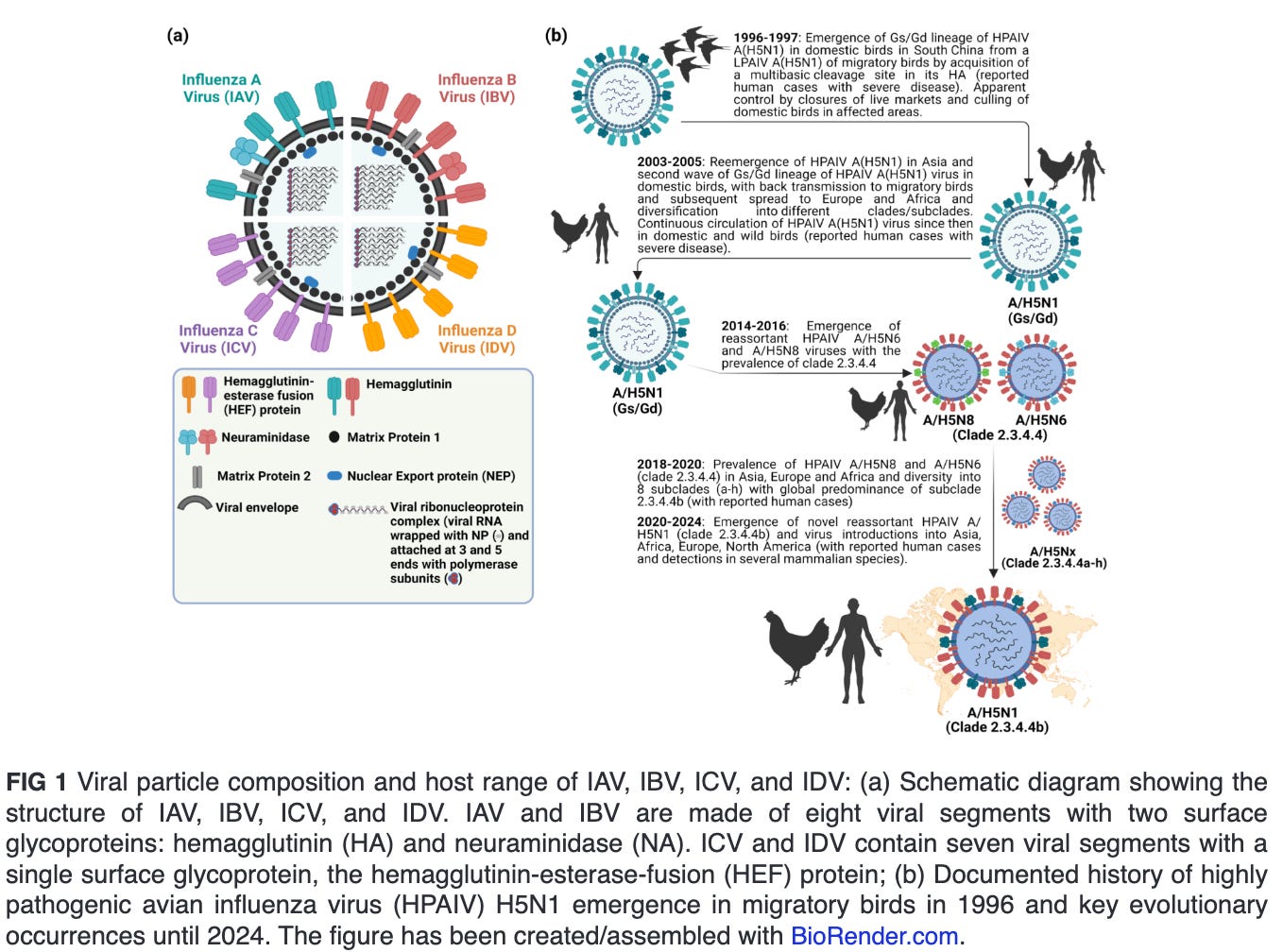
Influenza viruses are classified into four genera, namely Alphainfluenzavirus (influenza A virus [IAV]), Betainfluenzavirus (influenza B virus [IBV]), Gammainfluenzavirus (influenza C virus [ICV]) and Deltainfluenzavirus (influenza D virus [IDV]) (Fig. 1a). Seasonal epidemics in humans are caused annually due to infections with human IAV and IBV. In contrast to IBV, which is believed with some possible exceptions to be confined to humans, IAV is found to infect multiple animal species, including pigs, horses, marine mammals, dogs, bats, poultry, and several wild bird species. Infections with ICV could result in mild human illness, and it is not believed that they contribute to the main burden of severe respiratory disease in humans, as opposed to IAV and IBV. IDV infection occurs mainly in cattle, a natural reservoir with no reported cases in humans (1, 2). Nevertheless, evidence suggest that IDV can result in exposure of the workers due to occupational contact with infected cattle (3, 4). All influenza pandemics were caused by novel reassortants of IAV involving progenitor avian influenza viruses (AIV).
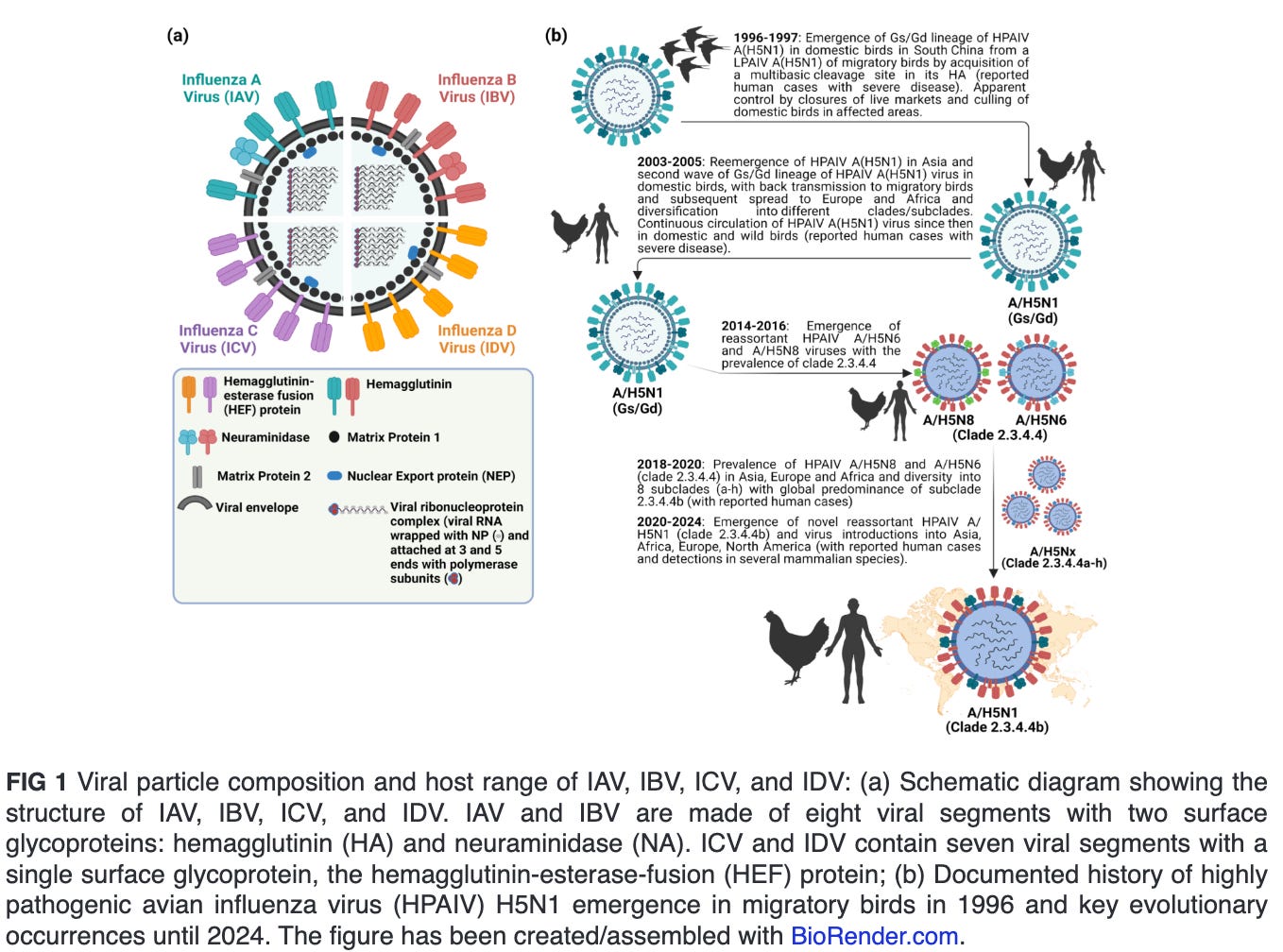
ECOLOGY AND EVOLUTION OF HPAIV H5N1 CLADE 2.3.4.4B
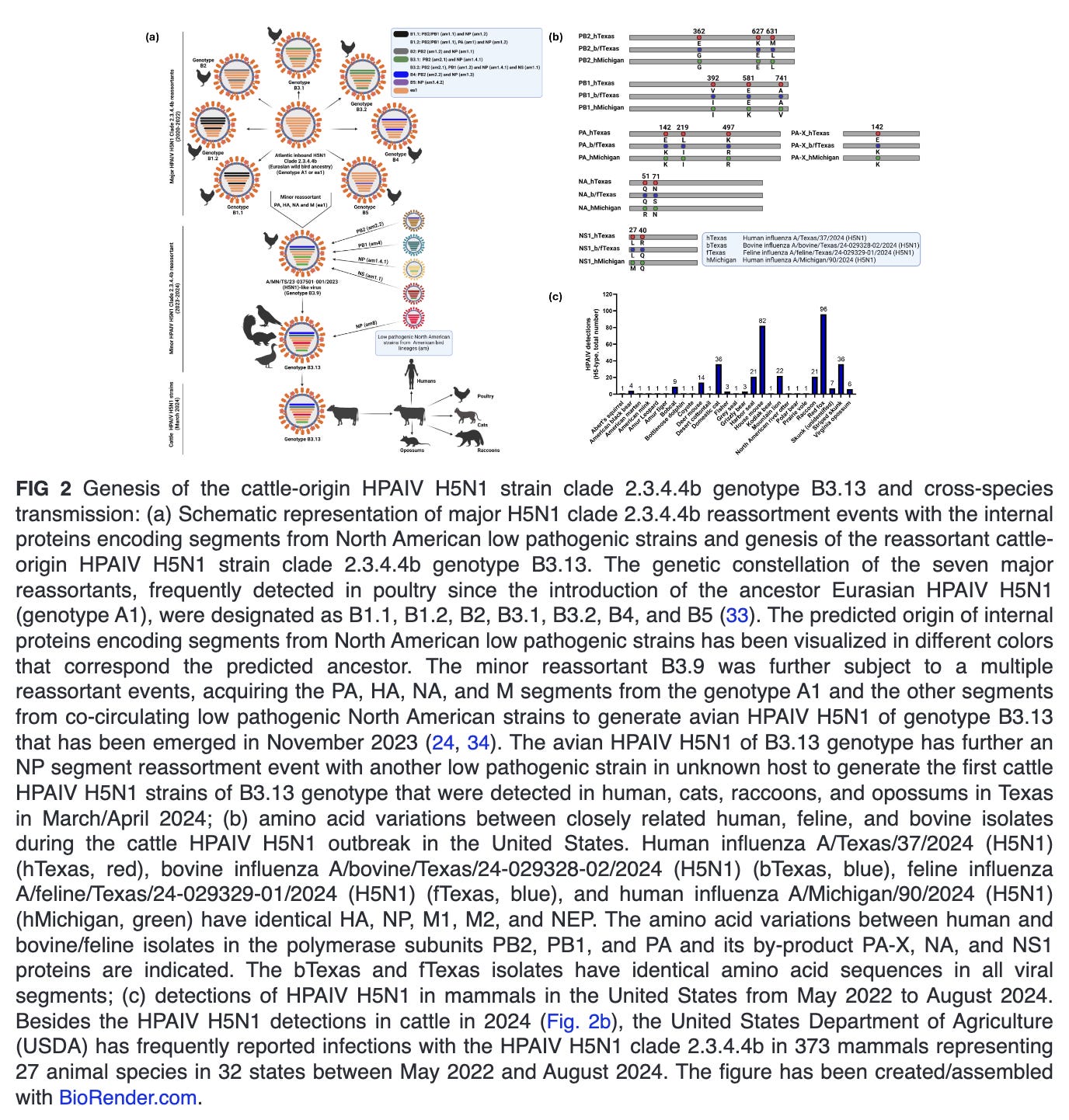
Since the emergence of highly pathogenic avian influenza virus (HPAIV) A/goose/Guangdong/1/1996 (Gs/Gd) H5N1 in China, the Gs/Gd lineage of HPAIV H5Nx has spread globally via migratory wild birds to infect various species and pose a threat to animal and human public health (1). Meanwhile, the H5-type Gs/Gd evolved into 10 distinct clades (0–9), and some of these clades are further categorized into different subclades (11). In late 2013, HPAIV H5N8 clade 2.3.4.4 emerged in China and shortly spread in early 2014 to South Korea and Japan (12). Later in 2014, the virus was further transmitted along different flyways to cause multiple outbreaks among poultry and wild birds in Asia, Europe, and North America (13). Meanwhile, the virus underwent multiple reassortment events with other AIV resulting in several H5Nx subtypes (e.g., H5N1, H5N2, H5N5, and H5N6) with eight subclades (2.3.4.4a–2.3.4.4h) (14–16). In 2020, a novel reassortant HPAIV H5N1 clade 2.3.4.4b emerged in the Middle East and became the most predominant reassortant/variant in Asia, Europe, and Africa by 2021 (17). In December 2021, the HPAIV H5N1 was further spread via migratory birds across the Atlantic Ocean from Europe to North America, leading to several outbreaks among poultry populations (Fig. 1b) (18). Shortly thereafter, the HPAIV H5N1 clade 2.3.4.4b has reached Central and South America and has been frequently reported in farmed animals, several mammals, including humans and marines, and outdoor animals (13, 19–21). This wide host range and multiple cross-species infections with HPAIV H5N1 in wildlife and domestic animals has been recently expanded following the ongoing and unprecedented epizootic in dairy cattle (cows). Except for minks, marine mammals, and cows, infections of mammals with HPAIV H5N1 have not been documented to result in a mammal-to-mammal transmission (22–24). This highlights the importance of the three animal species, especially dairy cows, as potential factors in the emergence of HPAIV H5N1 variants that may transmit easily among human populations.
Until recently, cattle have been considered unlikely hosts for HPAIV H5N1 due to significant differences in their respiratory tract physiology compared to birds. However, a recent study demonstrated that avian SA-α2,3-Gal-β1,4, and SA-α2,3-Gal-β1,3 are present in the mammary gland, respiratory tract, and cerebrum of beef and/or dairy cattle. This finding provides a rationale for the observed propensity of HPAIV H5N1 to infect and replicate in the mammary glands (25). As the virus does not appear to infect cows efficiently via the respiratory route, most of the evidence points to cow-to-cow transmission via using the same milking devices that, once contaminated with infected milk, can inoculate the virus to the mammary gland of a non-infected cow during the milking process (26). Missing in this scenario is how the virus entered for the first time the mammary tissue of a cow, but one could speculate that perhaps a milking device was contaminated with HPAIV H5N1 from an infected bird, initiating the chain of cow-to-cow transmission.
Since its emergence in late 2020, HPAIV H5N1 clade 2.3.4.4b has caused worldwide epizootics in Europe, Africa, Asia, and the Americas (27, 28), and it has diversified into several genotypes circulating in several bird species, and showed the ability to infect more than 30 mammalian species with sporadic human cases (29). The origin of HPAIV H5N1 clade 2.3.4.4b is traced back as novel reassortant between H5N8 clade 2.3.4.4b and other IAV subtypes (30). Remarkably, HPAIV H5N1 clade 2.3.4.4b demonstrated substantiable geographic spread and host adaptation, facilitating its spread across various continents via migratory bird routes. By late 2021, the Eurasian HPAIV H5N1 clade 2.3.4.4b had reached North America through wild birds and subsequently been detected in a variety of mammalian species.
In 2024, the first HPAIV H5N1 isolates from dairy cattle in Texas shared nearly identical genome sequences and were identified based on the phylogenetic analyses as clade 2.3.4.4b genotype B3.13, a reassortant of the minor B3.9 genotype reassortant of the Atlantic HPAIV H5N1 clade 2.3.4.4b (genotype A1) (24). This B3.13 genotype had emerged in late 2023 via reassortment between Eurasian wild bird H5N1 lineages (PA, HA, NA, and M gene segments) and American non-H5N1 wild bird lineages (PB2, PB1, NP, and NS gene segments) (Fig. 2a) (24, 31). Few weeks later, HPAIV H5N1 clade 2.3.4.4b genotype B3.13 was reported in several dairy farms in other localities of the United States, representing a multistate epizootic (32). Remarkably, the HPAIV H5N1 clade 2.3.4.4b genotype B3.13 reassortant virus implicated in the United States dairy cattle outbreak has never been detected so far in Europe. Similarly, no other Eurasian-North American reassortants have been recorded in Europe, despite detections of these viruses in Canada and the United States since 2014.
Different mutations in the HA and the polymerase subunits, which most likely facilitates viral entry and replication, respectively, in the new mammalian host cells, have been identified in dairy cattle HPAIV H5N1 (Table 1) (32). These mutations were found to enhance the binding affinity for sialic acid (SA) receptors prevalent in the mammalian upper respiratory tract, thereby enabling the virus to cross species and infect mammals. Additionally, alterations in the NA protein, which aids in the release of newly formed viral particles from infected host cells, have also been observed, most likely contributing to increased replication and transmission efficiency in cows (30–32). These changes might not only increase the ability of HPAIV H5N1 to establish infections in cattle but also may enhance its potential to spread from cattle to other mammals. This raises significant health concerns for both animals and humans.

What is the actual early history here? Where and when was H5N1 (avian influenza) first detected?

Note the author affiliations. The senior author is from the precise lab that the Hulscher et al paper asserts was the recent origin of the currently circulating H5N1.
Historically, the first identification of an AI virus in birds was an HPAI virus, termed fowl plague virus, which was initially identified in Northern Italy during the 1880s (Kaleta and Rulke 2008).
The fowl plague cases from the 1880s to 1959 were exclusively H7N7 and H7N1 HPAI viruses, but in 1959, the first H5 HPAI virus appeared in chickens in Scotland, H5N1 (Kaleta and Rulke 2008; Swayne et al. 2020). From the 1880s to 1959, fowl plague spread throughout Europe, Asia, and Africa and was reported from North and South America.
Since 1959, with well-maintained unique virus isolates in archives, outbreaks caused by HPAI viruses have mostly arisen from virulence shifts by LPAI viruses with limited spread and eradication from poultry via stamping out programs. The major exception has been the A/goose/Guangdong/1/1996 (Gs/GD) lineage of H5, which arose in 1996 and has spread and been maintained in poultry and wild aquatic bird reservoirs until the current date, producing infections in poultry, wild birds, or humans in 84 countries in Asia, Africa, Europe, and North America (Röhm et al. 1995; Swayne et al. 2020).
The mechanisms by which H5/H7 LPAI viruses change into HPAI virus include mutation, insertion, and recombination in the proteolytic cleavage site of the HA. The HA is the AI virus surface glycoprotein responsible to attach the virus to the host cell receptor to initiate the infection life cycle. After the virus enters the cell the new viruses start to be produced, including the inactive precursor HA protein (HA0). The HA0 is cleaved at the proteolytic cleavage site by cellular proteases of the host to form the functional subunits HA1 and HA2, resulting in an infectious virus. The cleavage site region in the HA0 consists of an arginine (R) residue adjacent to a conserved glycine (G) (Garten and Klenk 1983; Swayne et al. 2020). The number of basic amino acids at the HA cleavage site plays a critical role in virulence by determining which proteases cleave the HA0 and consequently in which cell types and tissues in the host the AI viruses can replicate (Bosch et al. 1981; Kawaoka et al. 1987; Horimoto and Kawaoka 1994; Swayne et al. 2020). First, the AI virus HA cleavage site can be classified as a monobasic (e.g., PEKQTR/GLF) or multibasic (e.g., PQRKKR/GLF). The monobasic cleavage site usually contains one or two nonconsecutive basic amino acids, arginine (R) or lysine (K), in the critical position and is cleaved by trypsin or trypsin-like proteases that confines virus replication in the epithelial cells of the respiratory and gastrointestinal tracts (Suarez 2016; Swayne et al. 2020). The H5/H7 LPAI viruses maintain the general principle of monobasic cleavage site, although in the wild birds, these subtypes have varying patterns of amino acids at the cleavage site (Suarez 2016). The H5 LPAI viruses usually encode the QRETR/G sequence, whereas H7 LPAI viruses typically possess the NPKTR/G sequence at the cleavage site. An increased number of basic amino acids and/or a lengthening of the proteolytic cleavage site to a minimum motif of four basic amino acids change the virus to high virulence by allowing the HA0 to be cleaved by several ubiquitous cellular proteases (furin-like proteases), which permit the virus to replicate in cells of multiple tissues, increasing the potential to cause a systemic disease and lethal infection in gallinaceous host (OIE 2019b; Swayne et al. 2020). More specifically, the changes observed in the HA cleavage site of HPAI viruses occurred because of (a) substitutions of nonbasic with basic amino acids and in some situations accompanied by loss of a shielding glycosylation site, (b) insertions of multiple basic amino acids from codons duplicated, (c) short inserts of basic and nonbasic amino acids from unknown source, or (d) nonhomologous recombination with cellular (e.g., host 28S RNA) or viral RNAs (e.g., RNA coding NP or M protein) that lengthen the proteolytic cleavage site. The changes (a), (b), and (c) have been observed in the H5/H7 HPAI viruses, whereas (d) has only been observed in H7 HPAI viruses (Suarez et al. 2004; Pasick et al. 2005; Maurer-Stroh et al. 2013; Swayne et al. 2020).
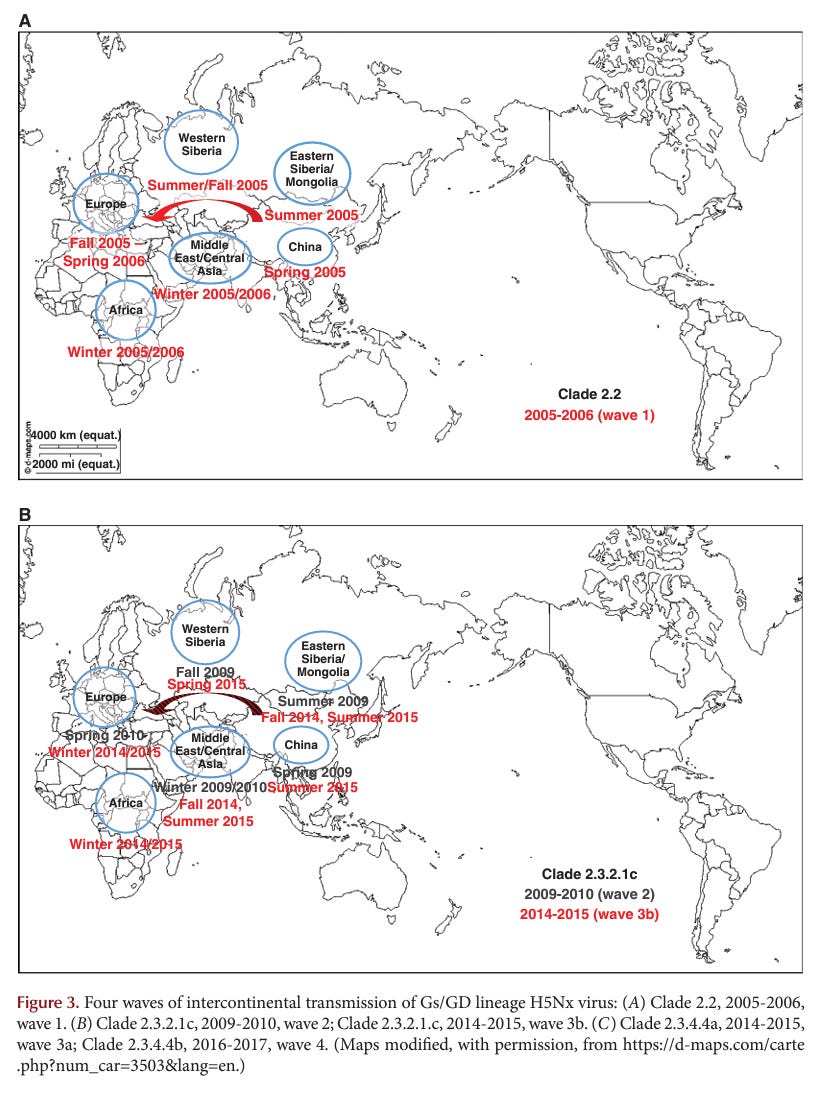
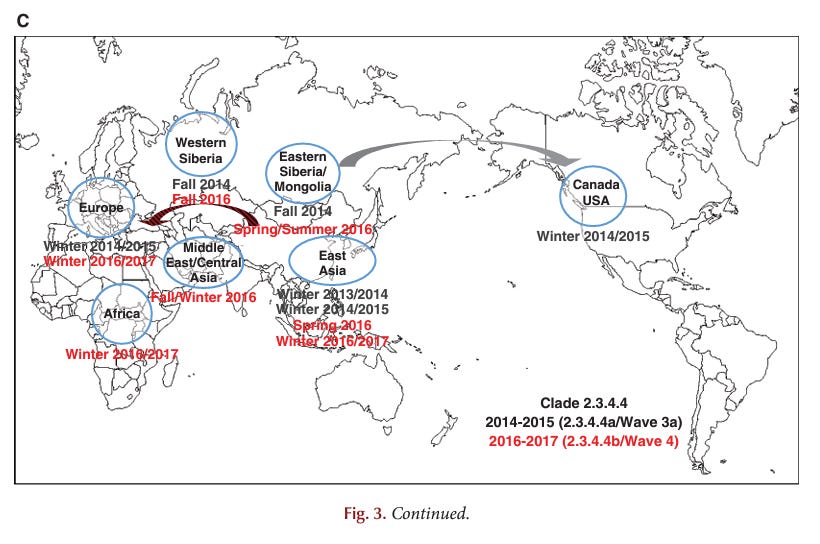
CONCLUDING REMARKS
Historically, the first AI virus in birds was identified in the 1880s as an HPAI virus (i.e., fowl plague virus). Forty-two documented epidemics or limited outbreaks of HPAI have occurred since the discovery of AI viruses as the cause of fowl plague in 1955. Since 1959 with well-maintained virus archives, most HPAI virus have arisen from changes in LPAI viruses with limited spread of outbreaks and eradication from poultry. However, the Gs/GD H5Nx, Mexican H7N3, and Chinese H7N9 HPAI viruses are concerning because these strains have not been eradicated and still cause poultry outbreaks. In particular, the Gs/GD H5Nx and Chinese H7N9 viruses pose a significant pandemic threat to human health. The Gs/GD lineage of H5 has a broad One Health impact as it continues to cause infections, disease, and death in poultry, wild birds, and humans. Furthermore, the Gs/GD lineage outbreak has affected more poultry and countries than the other 41 HPAI outbreaks combined.
Although chicken HPAI viruses are derived from LPAI viruses of aquatic wild birds, the emergence of HPAI virus does not always happen. The mechanisms by which H5/H7 HPAI virus has emerged is via mutation, insertion, or recombination within the HA—more specifically changes in the proteolytic cleavage site. The number of basic amino acids in the HA cleavage site plays a critical role in virulence determining which proteases can cleave HA and in which tissues the AI viruses can replicate. In general, the mechanisms for amino acid insertions in the cleavage site appear to be different in many cases, and for this reason the genetic basis to predict which LPAI viruses will result in the emergence of a HPAI virus remains unclear. The presence or absence of the N-linked glycans and key histidine residues in the HA are reported to influence the AI virus phenotype. In addition, changes in other AIV gene segments can increase or decrease the maximal phenotypic expression of the HA usually through increasing replication efficiency and release. In general, molecular techniques such as next-generation sequencing and reverse genetics have helped to identify changes associated with high virulence. However, significant emphasis on basic research is needed to understand why changes occur only in certain LPAI precursors that increase their pathogenicity, which, once understood, could enhance our ability to predict and subsequently prevent conversion events and potentially lead to a reduced number of HPAI outbreaks. With the ultimate goal of eradication, enhanced active surveillance, education, biosecurity, rapid molecular, and characterization efforts will provide the best opportunity for early detection and elimination of HPAI in domestic poultry.
So, was the current Bird Flu “Made in America”?
Lets recap:
Dr. Peter McCullough: "The current strain of bird flu is a product of gain of function research done in the USDA Poultry Research Laboratory in Athens, Georgia. So it is a man made problem that, our farms are experiencing right now. It's in the peer reviewed literature, and, I mean, it's really you know, the next steps in this outbreak is, for people to understand, you know, the ramifications of it personally and how to be prepared."
This statement is readily demonstrated to be false. It is also irresponsible, in my opinion. There is no evidence provided by Dr. McCullough or colleagues that the currently circulating “strain of bird flu is a product of gain of function research done in the USDA Poultry Research Laboratory in Athens, Georgia”. This is a hypothesis of Dr. McCullough and his two colleagues. The “peer reviewed” manuscript that Dr. McCullough cites does not in any way draw this conclusion, but instead uses terms such as “Origin may be… Suggest that laboratory activities… could have contributed… causation has not been established.”
Whether or not this paper should be retracted is up to the Editor of this obscure specialty journal. However, in my experience, in most journals, the failure to disclose a significant conflict of interest would be sufficient grounds for retraction.
Personally, I think that, at a minimum, an apology to the researchers at the Erasmus Institute and the National Poultry Research Center, Agricultural Research Service, U.S. Department of Agriculture, Athens, Georgia should be issued. These unfounded and widely publicized claims have damaged their reputations.

The problem with people making unsupported claims like this is that it delegitimizes the whole issue. The boy who cried wolf.
Thanks for your continued help separating the "wheat" of accurate information and analysis from all the "chaff" of misinformation and outright lies that make up what seems to be a majority of the information out in the public sector. I hope you and your family have a very prosperous and beneficial New Year.